

From: Stefan Gajewski
Date: 14 December 2011 04:47
I am looking at a highly unusual crystal lattice right now and can't figure out what is going on, so I decided to ask the experts.
I recently got data on a oligomeric protein with many highly correlated NCS units (4.0A resolution, linear R-sym is 0.16-0.21 in I4, I422, F222, C2 and 0.12 in P1) with severe anisotropic diffraction (according to diffraction anisotropy server, the F/sigma drops below 3 at a=6.1 b=6.1 c=never, suggested isotropic B-sharpening -125A^2) This lattice has a problem. The apparent unit cell is rather huge (roughly 180 180 620 / 90 90 90)
The unit cell dimensions are almost perfectly I4 and the presence of systematic absent reflections >50 I/s in I41 and I4122 suggest no screw axis. I used a very closely related structure solved at 4.2A as molecular replacement model and got a solution from the anisotropy corrected data in I422 space group with two oligomers in the asymmetric unit cell.
Confidence of the MR "solution" is quite high since (a)the MR replacement put one model one NCS raster off the "true" position resulting in a clash with the second one in an empty region of the map and additional electron density on the other side which corresponds perfectly to the wrongly positioned monomer, and (b) after rotating the model in the "right" position I could refine the structure to R-work=0.31. R-free=0.35 in one run of rigid body refinement followed by NCS restrained simulated annealing refinement (phenix.refine), which is in my opinion really good at such an early stage of refinement given the low overall resolution and even lower completeness of strong reflections in a and b due to high anisotropy (observables to atoms ratio is about 3:1) . I can even see clear density for some of the bulky sidechains which were not included in the model.
Now here is the baffling thing. The unit cell is almost empty with an apparent solvent content of >78%. The molecules cluster around the c-axis and at the origin with an empty gap in a and b of at least 15A and up to 165A(!) in the longest dimension. There is no sign of electron density that would indicate a missing protein in that region and ~98% of my model is already accounted for by the density in the 2Fo-Fc map, making a contact of disordered protein regions across the ASUs unlikely. In fact, the protein density is well defined at the closest gap and no mainchain atom is unaccounted for in that region. The oligomer has a magnitude of ~105A x 70A. I heavily doubt that a crystal lattice with such little contacts and holes as huge as these can exist and therefore think that:
(a) the R-factors are misleading me to think the solution is correct and complete
(b) I must have been doing something really wrong
Since proteins from this family have a well established history of producing twinned crystals I had a look at that possibility. Analyzing the anisotropy corrected I4 data for twinning (Padilla & Yeates method) revealed a 2-fold twin law with a twin fraction of 0.42 which would make the discrimination between an almost perfectly merohedral twin in I4 and a (non twinned ?) I422 extremely difficult (to me). MR with anisotropy corrected I4 data gave the same crystal packing and hence the same void solvent region. MR in lower point groups was not successful so far although I haven't pursued that idea vigorously. The same data in I422 has no indication for twinning and in C2 three 2-fold twin laws.
Anomalous data is not easily available since those crystals grow in about one year and getting another crystal is also not very likely because this IS "the other crystal".
I am clueless now on how to proceed here and would appreciate advice from experienced crystallographers on what to try first.
Am I worrying too much about the packing?
Is it even possible to have such an enormously huge solvent region in a protein crystal?
What is the recommended protocol when dealing with many and very strongly correlated NCS units, putative twinning and severe anisotropy all at the same time?
Stefan Gajewski
----------
From: Pavel Afonine
Hi Stefan,
----------
From: Bosch, Juergen
If you have air in the packing that's worrysome. If symmetry mates don't make crystal contacts you are in trouble.
Have you checked a simple selfrotation function in your currently favored space group ?
Do you have sufficient data collected to start out in P1 or C2 ? Then I would start there and systematically look at selfrotation functions in those space groups. Also check the native Patterson for translational NCS.
4 A is not great for stable refinement of cell parameters, which program did you use and which parameters did you fix?
Did you use main.ncs=true in the SA approach ?
Pointless or xtriage ?
Why does it take a year to grow those crystals ?
Out of curiosity, how did you collect on this crystal without overlapping reflections ?
Good luck,
Jürgen
......................
Jürgen Bosch
Date: 14 December 2011 04:47
I am looking at a highly unusual crystal lattice right now and can't figure out what is going on, so I decided to ask the experts.
I recently got data on a oligomeric protein with many highly correlated NCS units (4.0A resolution, linear R-sym is 0.16-0.21 in I4, I422, F222, C2 and 0.12 in P1) with severe anisotropic diffraction (according to diffraction anisotropy server, the F/sigma drops below 3 at a=6.1 b=6.1 c=never, suggested isotropic B-sharpening -125A^2) This lattice has a problem. The apparent unit cell is rather huge (roughly 180 180 620 / 90 90 90)
The unit cell dimensions are almost perfectly I4 and the presence of systematic absent reflections >50 I/s in I41 and I4122 suggest no screw axis. I used a very closely related structure solved at 4.2A as molecular replacement model and got a solution from the anisotropy corrected data in I422 space group with two oligomers in the asymmetric unit cell.
Confidence of the MR "solution" is quite high since (a)the MR replacement put one model one NCS raster off the "true" position resulting in a clash with the second one in an empty region of the map and additional electron density on the other side which corresponds perfectly to the wrongly positioned monomer, and (b) after rotating the model in the "right" position I could refine the structure to R-work=0.31. R-free=0.35 in one run of rigid body refinement followed by NCS restrained simulated annealing refinement (phenix.refine), which is in my opinion really good at such an early stage of refinement given the low overall resolution and even lower completeness of strong reflections in a and b due to high anisotropy (observables to atoms ratio is about 3:1) . I can even see clear density for some of the bulky sidechains which were not included in the model.
Now here is the baffling thing. The unit cell is almost empty with an apparent solvent content of >78%. The molecules cluster around the c-axis and at the origin with an empty gap in a and b of at least 15A and up to 165A(!) in the longest dimension. There is no sign of electron density that would indicate a missing protein in that region and ~98% of my model is already accounted for by the density in the 2Fo-Fc map, making a contact of disordered protein regions across the ASUs unlikely. In fact, the protein density is well defined at the closest gap and no mainchain atom is unaccounted for in that region. The oligomer has a magnitude of ~105A x 70A. I heavily doubt that a crystal lattice with such little contacts and holes as huge as these can exist and therefore think that:
(a) the R-factors are misleading me to think the solution is correct and complete
(b) I must have been doing something really wrong
Since proteins from this family have a well established history of producing twinned crystals I had a look at that possibility. Analyzing the anisotropy corrected I4 data for twinning (Padilla & Yeates method) revealed a 2-fold twin law with a twin fraction of 0.42 which would make the discrimination between an almost perfectly merohedral twin in I4 and a (non twinned ?) I422 extremely difficult (to me). MR with anisotropy corrected I4 data gave the same crystal packing and hence the same void solvent region. MR in lower point groups was not successful so far although I haven't pursued that idea vigorously. The same data in I422 has no indication for twinning and in C2 three 2-fold twin laws.
Anomalous data is not easily available since those crystals grow in about one year and getting another crystal is also not very likely because this IS "the other crystal".
I am clueless now on how to proceed here and would appreciate advice from experienced crystallographers on what to try first.
Am I worrying too much about the packing?
Is it even possible to have such an enormously huge solvent region in a protein crystal?
What is the recommended protocol when dealing with many and very strongly correlated NCS units, putative twinning and severe anisotropy all at the same time?
Stefan Gajewski
----------
From: Pavel Afonine
Hi Stefan,
1)
just out of curiosity I wrote a tiny script using CCTBX that estimates solvent content via bulk-solvent mask, and quickly run this script for all PDB structures for which I could re-calculate the R-work within 5% from published value. Also, this script extracted the solvent content values reported in PDB file header. Here is what I get:
Histogram of solvent contents (estimated via mask):
Solvent content Number of structures
5.980 - 14.482 : 11
14.482 - 22.984 : 109
22.984 - 31.486 : 396
31.486 - 39.988 : 3590
39.988 - 48.490 : 11442
48.490 - 56.992 : 11707
56.992 - 65.494 : 6524
65.494 - 73.996 : 2561
73.996 - 82.498 : 510
82.498 - 91.000 : 19
Histogram of solvent contents (extracted from REMARK records):
Solvent content Number of structures
6.000 - 14.300 : 91
14.300 - 22.600 : 550
22.600 - 30.900 : 2046
30.900 - 39.200 : 6487
39.200 - 47.500 : 9566
47.500 - 55.800 : 9050
55.800 - 64.100 : 5853
64.100 - 72.400 : 2420
72.400 - 80.700 : 720
80.700 - 89.000 : 86
So, your 78% is not that uncommon although it is at the high(ish) end.
2) Does Xtriage suggest twinning? If so what happens if you refine with the twin law?
3) Make sure you look at both, 2mFo-DFc with and without missing Fobs filled with DFc (depending on completeness of your data that may make a big difference).
Pavel
----------
From: Bosch, Juergen
If you have air in the packing that's worrysome. If symmetry mates don't make crystal contacts you are in trouble.
Have you checked a simple selfrotation function in your currently favored space group ?
Do you have sufficient data collected to start out in P1 or C2 ? Then I would start there and systematically look at selfrotation functions in those space groups. Also check the native Patterson for translational NCS.
4 A is not great for stable refinement of cell parameters, which program did you use and which parameters did you fix?
Did you use main.ncs=true in the SA approach ?
Pointless or xtriage ?
Why does it take a year to grow those crystals ?
Out of curiosity, how did you collect on this crystal without overlapping reflections ?
Good luck,
Jürgen
......................
Jürgen Bosch
----------
From: Phil Evans
Don't forget that if you have poorly resolved spots due to the long axis, the intensity statistics may falsely seem to indicate twinning, since weak spots may be contaminated by neighbouring strong ones
Phil
----------
From: Tjaard Pijning
Stefan,
Just to relieve some of your worries about packing... recently I've had two cases of crystal lattices with high solvent content and huge solvent channels along one of the axes :
- P 3(1)21, a = b= 174 A, c = 78 A, solvent content = 85% with channels of 140 A
- P 6(2)22, a = b = 184 A, c = 374 A, solvent content = 85% with channels of 100 A
You can see for yourself in the pictures attached.
Diffraction was in the 3-4 A range and somewhat anisotropic but both structures were solved easily by MR using high-resolution homologous structures.
Kind regards,
Tjaard
----------
From: Stefan Gajewski
Jürgen,
Yes, both selfrotation function and patterson map do not look suspicious in I422.
I got ~220 degrees before the radiation damage became significant, so there should be enough data and I will look into it.
HKL2000 without fixing parameters. mosflm can't hold on the lattice and I haven't tried d*trek, yet.
yes
Pointless or xtriage ?
xtriage
Well, other crystals don't diffract and the protein is quite stable in solution.
That's how it is, I guess?
I got away with 1 second exposure and 0.75 degree oscillation at 650mm detector distance. there are some predicted overlaps but they are in those regions that are empty due to anisotropy. Our cryo condition gives well separated small spots of nice, round shape, mosaicity is ~0.6. The pattern itself looks great, although the beamstop shadow is quite big on the frames.
Thank You,
Stefan
----------
From: Bosch, Juergen
Give XDS a try with your data or d*trek with 3d profile fitting.
Did you try iMosflm or the old Mosflm ? If old, then POSTREF WIDTH 15 might help and POSTREF FIX BEAM
You saw the nice packing from Tjaard, if all your molecules have contact with each other than that's fine I was just concerned about molecules in space without contact, then you are missing something.
Still reprocessing the data (sorry Wladek) might squeeze out a bit more of your existing data. In particular if you use NCS averaging with those weak highres reflections you might get better side chain density.
Jürgen
......................
Jürgen Bosch
Johns Hopkins University
Jürgen Bosch
Johns Hopkins University
----------
From: Debasish Chattopadhyay
How about plotting the solvent content along with resolution limits of the structures?
Hi Stefan,
----------
From: Stefan Gajewski
Thanks to everyone so far.
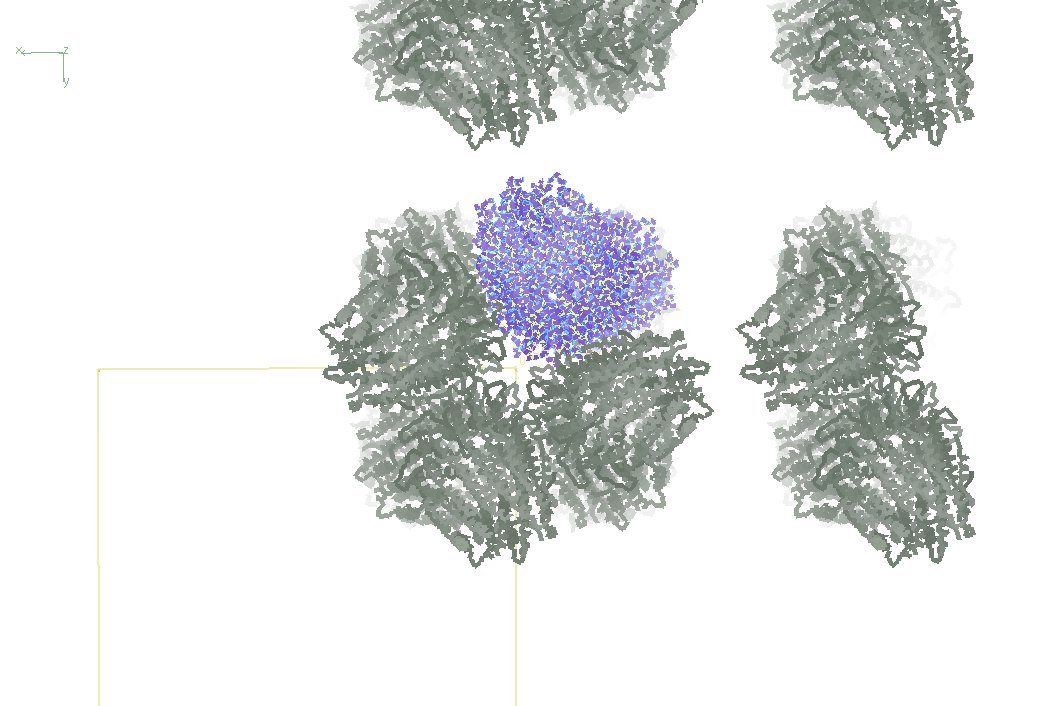
Here is a snapshot looking down the long dimension so you see the (absence) of contacts in a and b.
As I posted earlier, there is no mainchain atom in my protein unaccounted for in that region of the lattice. the few disordered elements are far away from that close gap.
And yes, I use NCS restraints.
Stefan
----------
From: Pavel Afonine
Hi Debasish,
here it is:
Resolution Number of Solvent % (mean/min/max) Solvent % (mean/min/max)
range structures from remarks from mask
0.0-1.0 148 (37.3 9.9 61.7) (30.7 9.0 55.0)
1.0-1.5 2934 (44.6 6.0 75.0) (37.4 9.0 77.0)
1.5-2.0 14270 (48.7 16.4 81.1) (43.6 8.0 77.0)
2.0-2.5 11432 (52.6 16.9 84.0) (50.8 7.0 83.0)
2.5-3.0 5467 (57.0 27.0 86.0) (57.5 25.0 88.0)
3.0-3.5 1140 (61.3 20.0 89.7) (62.6 30.0 89.0)
3.5-4.0 151 (65.9 35.0 85.0) (67.1 45.0 88.0)
4.0-9.0 27 (66.2 43.0 84.4) (67.4 50.0 81.0)
Pavel
----------
From: <Herman.Schreuder
Dear Stefan,
We recently had a case where a protein mutant formed tetramers with internal 21212 symmetry. It turned out that these internal symmetry axis would align perfectly with the crystallographic symmetry axes, but with some translations. All processing programs including XDS and pointless would insists that the space group was P21212, as was wildtype. However, molecular replacement did not work, resulting in large gaps in the packing, or if one would push very hard, some well defined molecules and some poorly defined molecules.
You may have the same problem. The tetramers of octamers (I cannot deduce them from your picture) will have a perfect 4-fold or 422 symmetry, with will leave a very strong imprint on your diffraction data, like in our case. We solved to problem by going back to P1, and from the P1 solution deduce the real space group (which turned out to be P21). In your case I would process the data in P1 and do an MR search with the tetramers (or octamers) you show in your picture. The number of octamers to be found should be reasonable.
Best,
Herman
No comments:
Post a Comment