With TLS

Without TLS
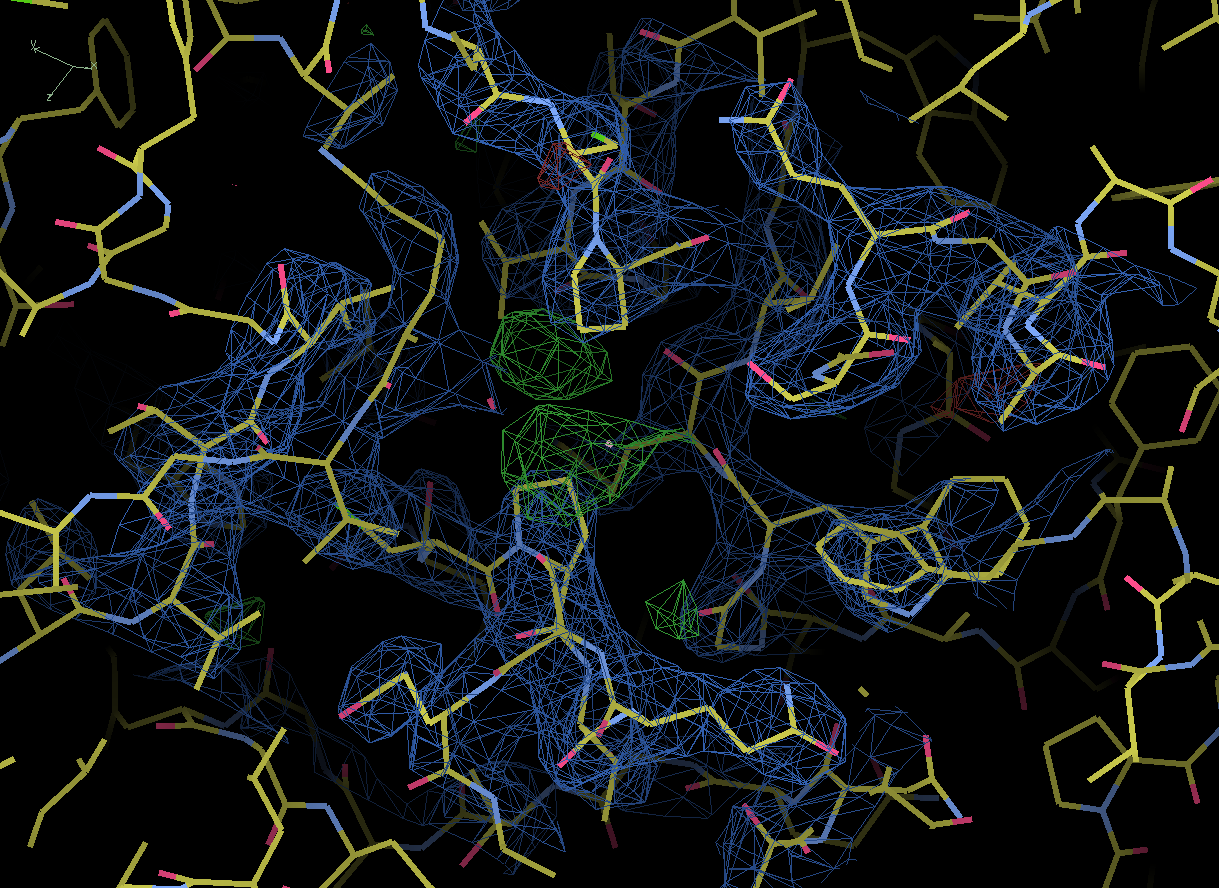
From: Naveed A Nadvi
Date: 19 February 2012 01:36
Dear crystallographers,
I am fairly new in crystallographic work and structure determination, but I thought this would be the best place to post my questions. We had collected structural data for a protein that diffracted to 3 A. We had used a previously deposited structure (1.5 A) for molecular replacement. Our final structure used NCS restraints refinement over 4 chains within the assymetric unit. We were able to assign some water moleules using COOT and subsequently removed 'bad waters' manually. We used automated settings when dealing with these water molecules. In all cases these water molecules were found in the same positions as the initial structure (1.5 A) that we had used as a search model. This gave us confidence in the placement of our water molecules. Finally we had run validation tools (MolProbity) and our structure was found to be with Molprobity score within the 100th percentile.
We then performed a TLS refinement (from TLSMD) to further improve R values. We used the final MolProbity-validated structure using 8 TLS groups and using PureTLS with constant B factor (50). We are observing large positive densities from the subsequent REFMAC5 refinement that are otherwise not observed in the absence of TLS refinement. My questions are:
1) Is TLS suitable for our dataset (3 A)?
2) Is TLS refinement independent of NCS refinement or should I define my NCS based on the 8 TLS groups?
3) Is it normal to see extra positive density after TLS refinement and what does it mean?
4) We had PEG4000 and Tris in our crystallization buffer. Could these 'blobs' represent these molecules or short water chains? I have attached images of the largest blob.
Any comments and suggestions would be highly appreciated.
Kind regards,
Naveed A Nadvi
----------
From: Nian Huang
MolProbability score doesn't mean too much in your case, since you are essentially using a 1.5 A model against a 3 A database. The differences in the blobs might caused by the different delta sigma settings when you were viewing these two models.
I have successfully used TLS for a 3 A dataset before. The blobs mean the discrepancies between you model and your data, no matter you are using TLS or not. If TLS doesn't give you better statics and map density, I would leave it out or change the TLS setting.
I wouldn't put any molecule in the densities you provided, as the model looks already congested enough from the angle I see. I might be wrong w/o the information of map setting.
Best,
Nian
----------
From: Ethan Merritt
On Saturday, 18 February 2012, Naveed A Nadvi wrote:
> Dear crystallographers,
>
>
>
> I am fairly new in crystallographic work and structure determination, but I thought this would be the best place to post my questions. We had collected structural data for a protein that diffracted to 3 A. We had used a previously deposited structure (1.5 A) for molecular replacement. Our final structure used NCS restraints refinement over 4 chains within the assymetric unit. We were able to assign some water moleules using COOT and subsequently removed 'bad waters' manually. We used automated settings when dealing with these water molecules. In all cases these water molecules were found in the same positions as the initial structure (1.5 A) that we had used as a search model. This gave us confidence in the placement of our water molecules. Finally we had run validation tools (MolProbity) and our structure was found to be with Molprobity score within the 100th percentile.
>
>
>
> We then performed a TLS refinement (from TLSMD) to further improve R values. We used the final MolProbity-validated structure using 8 TLS groups and using PureTLS with constant B factor (50). We are observing large positive densities from the subsequent REFMAC5 refinement that are otherwise not observed in the absence of TLS refinement.
Is it possible that the peaks are not higher in terms of absolute electron level,> Dear crystallographers,
>
>
>
> I am fairly new in crystallographic work and structure determination, but I thought this would be the best place to post my questions. We had collected structural data for a protein that diffracted to 3 A. We had used a previously deposited structure (1.5 A) for molecular replacement. Our final structure used NCS restraints refinement over 4 chains within the assymetric unit. We were able to assign some water moleules using COOT and subsequently removed 'bad waters' manually. We used automated settings when dealing with these water molecules. In all cases these water molecules were found in the same positions as the initial structure (1.5 A) that we had used as a search model. This gave us confidence in the placement of our water molecules. Finally we had run validation tools (MolProbity) and our structure was found to be with Molprobity score within the 100th percentile.
>
>
>
> We then performed a TLS refinement (from TLSMD) to further improve R values. We used the final MolProbity-validated structure using 8 TLS groups and using PureTLS with constant B factor (50). We are observing large positive densities from the subsequent REFMAC5 refinement that are otherwise not observed in the absence of TLS refinement.
but only in terms of RMSD? That is, if the TLS treatment cleans up the map
everywhere, then whatever peaks are left will deviate more from the (now lower)
mean value even though their absolute size is the same.
In other words, the "3 sigma" contours in your first map may be more like
6 sigma contours in your second (cleaner) map.
> My questions are:
>
> 1) Is TLS suitable for our dataset (3 A)?
Certainly TLS can help a lot at 3A for some structures. In general the more
anisotropy is present, the more it helps to include it in your model somehow -
and TLS is a "cheap" way to include it in your model. But if your structure does
not have much anisotropy, then adding TLS to describe it won't have much effect.
> 2) Is TLS refinement independent of NCS refinement or should I define my NCS based on the 8 TLS groups?
> 3) Is it normal to see extra positive density after TLS refinement and what does it mean?
Ethan
----------
From: Naveed Ahmed Nadvi
Dear Crystallographers,
Thank you for your responses. The density map levels were 0.11e/A^3 (3.00 A) for both images with and without TLS refinement. When I superposed the deposited structure I could see the extra density was due to water moleucles that were seen in the higher resolution deposited structure. It is so interesting how I could not see them in my data without doing TLS.
Performing the TLS refinement improved overall parameters:
no TLS/TLS
R 0.2425/0.2334
R-free 0.2809/0.2748
RMS BondLen 0.0092/0.0074
RMS BondAngle 1.1812/1.1407
ChirVol 0.0806/0.0779
My question is, do the positive density seen after TLS refinement justify adding these solvent molecules especially when they were not observed without TLS refinement?
Thank you once again for your insights!
Naveed
----------
From: Horacio Botti
Dear Naveed A Nadvi
I think that your results highlight the fact that modelling the disorder/complex ordering of your crystal is relevant and in general, that we should take care in optimizing B factor refinement as a strong factor for model improvement.
In this sense I would not relay in one TLS group definition, even though you have obtain better results comparing no TLS vs TLS. Try common sense definitions also: does the protein have a hinge between two domains, use the domains as TLS groups, etc. Then, look for optimal NCS between TLS groups also.
Best wishes
Horacio
No comments:
Post a Comment