From: Kayashree M
Date: 19 October 2011 11:36
Dear users,
We have a structure which is a homodimer in the asymmetric unit.
PISA predicts most probable assembly as a dimer but this
dimeric assembly is different from what is solved (offcourse
we can generate the symmetry equivalent molecule and get that).
My question is - is it necessary that we deposit a structure, which
PISA predicted as most probable assembly in PDB as an
asymmetric unit & biological assembly or can we deposit a dimer
(asymmetric unit) and give explanation for the biological assembly
according to what PISA predicted.
Other than such predictions what other criteria needs to be
considered to say that one specific assembly is a biological assembly?
Another question-
In this case one of the chain has 3 MSE residues, while the other
chain has only 2 MSE (When we change MET to MSE, there will be a
huge negetive density coming up).
Are there any such instances in PDB, where two homodimer (or any mer)
will have different percentage of MSE?
Thanking you
With Regards
kavya
--
----------
From: Frederic VELLIEUX
Hi,
What you must deposit is what is present in the asymmetric unit of the crystal. The HEADER cards (and the publication) can describe the most likely biological assembly.
Why is that: there is no reason why the crystal should conform to the biological function (and associated quaternary structure), there are examples of having the
asymmetric unit different than the biological assembly. Such as a crystal asymmetric unit containing half a viral capsid.
So the PDB files contain "what you see" in the crystal, and there are places for the interpretation, such as these pictures appearing on the web page for the structure
showing the most likely biological unit. Or if you (as a user) request the PDB to provide you with the coordinates for that most probable biological assembly.
Fred.
----------
From: <eugene.krissinel
Dear Kavya,
If I understand your question correctly, it is about the choice of asymmetric unit for your deposition. In case of dimeric asymmetric unit (ASU), there are, indeed, a few valid possibilities and you arrive to just one of them in the course of structure solution. What you decide to deposit, is your own choice, and I would think that the PDB can suggest an alternative but they would not really insist on it (hopefully).
In case when ASU has the same multiplicity (number of chains) as the probable biological assembly, the latter is an ASU as well. In such a case, the PDB suggests to choose ASU in the form of that assembly, purely for simplicity. It seems to me that this is not an unreasonable suggestion and it would be nice if that were a common practice. I do not want to imply here that PISA will always make a correct prediction, therefore, one should always be critical and use as much experimental evidence as possible. However, deposition of biological assembly as ASU, where possible, is always preferential irrespectively of whether the assembly agrees with PISA predictions or not. Preferential does not mean mandatory though :)
I hope this helps,
Eugene.
----------
From: Ed Pozharski
As others have said - you are free to deposit whatever model you believe
is consistent with your experimental data. If we assume that you know
what the biological assembly is (see below), you still may (and people
do that) deposit a different arrangement - it's not wrong per se, but
think about non-structural folk (there should be a better word) that
will look at your deposited model and may not have the experience to
realize what they are looking at.
1. You need to show that your protein is a dimer in solution.
Gel-filtration (sometimes problematic), dynamic light scattering (often
problematic), analytical ultracentrifugation (less problematic but
instruments are not widely available) as well as methods I mention in 2
are useful here.
2. You need to show somehow that the dimer you see in crystal is the
same as dimer in solution. Many approaches are available here - I am a
recent SAXS convert, and I wholeheartedly recommend that, but your
mileage may vary. SAXS will also unequivocally determine the
oligomerization state. Cross-linking and HD exchange, both in line of
mass-spectrometry, are good ways to get it done too.
With that said, you may be able to get away with just the crystal
structure assuming that the PISA results are convincing (e.g. you have
one heavily hydrophobic interface that is large enough and much larger
than any other more polar alternatives.
This is rather curious. While it's definitely possible that your
protein prep had variable incorporation ratio, the fact that you see it
in a specific spot seems to suggest that selenium slightly alters the
structure directing the crystallization.
Hopefully someone knows the answer, but you can always just mine the
database.
Cheers,
Ed.
--
"I'd jump in myself, if I weren't so good at whistling."
Julian, King of Lemurs
----------
From: Kayashree M
Dear users,
Thank you all for the suggestions.
With Regards
Kavya
----------
From: James Stroud
----------
From: Bostjan Kobe
On 19/10/11 9:19 PM, "eugene.krissinel"
Maybe I am misunderstanding what you are saying Eugene, but the ASU
interface may not necessarily be the biological interface. I think it is
perfectly possible that two subunits in a biological dimer, for example,
may be related by crystallographic axis, but the NCS may be a different
non-physiological crystal contact, therefore the ASU won't be the same as
the biological dimer. So I am not sure if the above applies in general.
Bostjan
Bostjan Kobe
NHMRC Research Fellow
Professor of Structural Biology
School of Chemistry and Molecular Biosciences
and Institute for Molecular Bioscience (Division of Chemistry and
Structural Biology) and Centre for Infectious Disease Research
Cooper Road
University of Queensland
Brisbane, Queensland 4072
Australia
>
----------
From: Kayashree M
Respected Sir,
The space group is H3. if I generate the symmetry,
it appears to be a dimer of trimers stacked one above
the other with a rotation of 60 deg wrt each other, like
this - A1, A2, A3 (in one trimer) stacked upon
B1, B2, B3 (second trimer). So structure that is in the ASU
is with A1-B1 while PISA predicts A1-B2.
Thank you
With Reagrds
Kavya
----------
From: Vellieux Frederic
Hi,
If, in your case, no possible asymmetric unit can contain A1-B2, then you deposit A1-B1 (or I suppose A2-B2...) and indicate to the PDB (like placing cards in the header cards) the operator to be used (and the subunit it applies to) in order to generate the most likely biological dimer. Normally the PDB can take care of that.
Fred.
Kayashree M wrote:
From: Kayashree M
Thank you Sir for the suggestions.
----------
From: Eleanor Dodson
I am sure someone has suggested this - you can submit your coordinates to the PISA server which will give you a detailed analysis of contacts, then you choose which grouping you think makes the most reasonable assemblyn and take the asymmetric unit from that .
I actually like to do that while still refining - it makes coot a lot easier to use if you are working with a compact molecule!
Eleanor
----------
From: Kayashree M
Respected Madam,
Actually I was not aware of the fact that we have to deposit the structure which is most probable
biological assembly (since in PDB there are seperate representations for the asymmetric unit and
biological unit).
Also Since I dint have any experimental evidences other than PISA predictions and comparison
with the structures from different organisms, I continues to refine the model PHASER predicted.
In addition the PHASER refined model has 3 Cadmium ions inbetween the monomers strongly
coordinated by both the monomers. Hence I did not have any doubts upon this assembly. Finally
I submitted this assembly itself. and by mistake I mentioned most probably biological assembly
section while submitting the PDB, Which gave rise to all confusions.
Thanking you
With Regards
M. Kavyashree
----------
From: <eugene.krissinel
Dear Bostjan,
Not at all. But if anticipated biological assembly has the same size as ASU,
then one should be able to choose ASU such that it is biological assembly.
Consider that, when there is more than 1 chain in ASU, its configuration
may be chosen in many different ways (but not completely arbitrarily,
of course). E.g., in case of heterodimeric ASU, _any_ hetero-pair of molecules
in crystal is a valid ASU (even if they do not make a contact :)). By this I mean
that, by applying all crystal symmetry operations and unit cell translations,
one can reconstruct the whole crystal from an arbitrary hetero-pair. In case
of homodimers it is slightly more complex, e.g. one has to exclude
monomers in parallel orientations.
I would think (am I wrong here?) that NCS-reduced PDB entries do not
represent crystal's ASU (simply because they are reduced by
_non-crystallographic_ symmetry operations). In order to get the ASU,
one needs to apply NCS to the content of the entry. E.g., PDB entry
1stm has 5 chains, but they do not make an ASU: applying all space
group symmetry operations would not reconstruct the crystal.
NCS-expansion, however, gives a 60-chain viral capsid, which is the
ASU. For fun, you may state that "your" ASU in this case is 2
complementing parts of neighbouring capsids, and this would be
"crystallographically correct". It is a matter of common sense (and
sense of beauty perhaps) that we choose to deposit a complete
viral capsid as an ASU here.
Eugene.
----------
From: James Stroud
----------
From: Kayashree M
Date: 19 October 2011 11:36
Dear users,
We have a structure which is a homodimer in the asymmetric unit.
PISA predicts most probable assembly as a dimer but this
dimeric assembly is different from what is solved (offcourse
we can generate the symmetry equivalent molecule and get that).
My question is - is it necessary that we deposit a structure, which
PISA predicted as most probable assembly in PDB as an
asymmetric unit & biological assembly or can we deposit a dimer
(asymmetric unit) and give explanation for the biological assembly
according to what PISA predicted.
Other than such predictions what other criteria needs to be
considered to say that one specific assembly is a biological assembly?
Another question-
In this case one of the chain has 3 MSE residues, while the other
chain has only 2 MSE (When we change MET to MSE, there will be a
huge negetive density coming up).
Are there any such instances in PDB, where two homodimer (or any mer)
will have different percentage of MSE?
Thanking you
With Regards
kavya
--
----------
From: Frederic VELLIEUX
Hi,
What you must deposit is what is present in the asymmetric unit of the crystal. The HEADER cards (and the publication) can describe the most likely biological assembly.
Why is that: there is no reason why the crystal should conform to the biological function (and associated quaternary structure), there are examples of having the
asymmetric unit different than the biological assembly. Such as a crystal asymmetric unit containing half a viral capsid.
So the PDB files contain "what you see" in the crystal, and there are places for the interpretation, such as these pictures appearing on the web page for the structure
showing the most likely biological unit. Or if you (as a user) request the PDB to provide you with the coordinates for that most probable biological assembly.
Fred.
----------
From: <eugene.krissinel
Dear Kavya,
If I understand your question correctly, it is about the choice of asymmetric unit for your deposition. In case of dimeric asymmetric unit (ASU), there are, indeed, a few valid possibilities and you arrive to just one of them in the course of structure solution. What you decide to deposit, is your own choice, and I would think that the PDB can suggest an alternative but they would not really insist on it (hopefully).
In case when ASU has the same multiplicity (number of chains) as the probable biological assembly, the latter is an ASU as well. In such a case, the PDB suggests to choose ASU in the form of that assembly, purely for simplicity. It seems to me that this is not an unreasonable suggestion and it would be nice if that were a common practice. I do not want to imply here that PISA will always make a correct prediction, therefore, one should always be critical and use as much experimental evidence as possible. However, deposition of biological assembly as ASU, where possible, is always preferential irrespectively of whether the assembly agrees with PISA predictions or not. Preferential does not mean mandatory though :)
I hope this helps,
Eugene.
----------
From: Ed Pozharski
As others have said - you are free to deposit whatever model you believe
is consistent with your experimental data. If we assume that you know
what the biological assembly is (see below), you still may (and people
do that) deposit a different arrangement - it's not wrong per se, but
think about non-structural folk (there should be a better word) that
will look at your deposited model and may not have the experience to
realize what they are looking at.
1. You need to show that your protein is a dimer in solution.
Gel-filtration (sometimes problematic), dynamic light scattering (often
problematic), analytical ultracentrifugation (less problematic but
instruments are not widely available) as well as methods I mention in 2
are useful here.
2. You need to show somehow that the dimer you see in crystal is the
same as dimer in solution. Many approaches are available here - I am a
recent SAXS convert, and I wholeheartedly recommend that, but your
mileage may vary. SAXS will also unequivocally determine the
oligomerization state. Cross-linking and HD exchange, both in line of
mass-spectrometry, are good ways to get it done too.
With that said, you may be able to get away with just the crystal
structure assuming that the PISA results are convincing (e.g. you have
one heavily hydrophobic interface that is large enough and much larger
than any other more polar alternatives.
This is rather curious. While it's definitely possible that your
protein prep had variable incorporation ratio, the fact that you see it
in a specific spot seems to suggest that selenium slightly alters the
structure directing the crystallization.
Hopefully someone knows the answer, but you can always just mine the
database.
Cheers,
Ed.
--
"I'd jump in myself, if I weren't so good at whistling."
Julian, King of Lemurs
----------
From: Kayashree M
Dear users,
Thank you all for the suggestions.
With Regards
Kavya
----------
From: James Stroud
This last sentence is a bit vague. Can you take the just dimer that PISA predicts, fit this dimer to the lattice (i.e. each monomer sitting correctly in density but retaining the dimeric relationship predicted by PISA), and then generate the complete lattice using just this fitted dimer and crystallographic symmetries?
If so, that means that the PISA dimer is equivalent to the ASU you can deposit the PISA dimer as the ASU.
James
----------
From: Bostjan Kobe
On 19/10/11 9:19 PM, "eugene.krissinel"
Maybe I am misunderstanding what you are saying Eugene, but the ASU
interface may not necessarily be the biological interface. I think it is
perfectly possible that two subunits in a biological dimer, for example,
may be related by crystallographic axis, but the NCS may be a different
non-physiological crystal contact, therefore the ASU won't be the same as
the biological dimer. So I am not sure if the above applies in general.
Bostjan
Bostjan Kobe
NHMRC Research Fellow
Professor of Structural Biology
School of Chemistry and Molecular Biosciences
and Institute for Molecular Bioscience (Division of Chemistry and
Structural Biology) and Centre for Infectious Disease Research
Cooper Road
University of Queensland
Brisbane, Queensland 4072
Australia
>
----------
From: Kayashree M
Respected Sir,
The space group is H3. if I generate the symmetry,
it appears to be a dimer of trimers stacked one above
the other with a rotation of 60 deg wrt each other, like
this - A1, A2, A3 (in one trimer) stacked upon
B1, B2, B3 (second trimer). So structure that is in the ASU
is with A1-B1 while PISA predicts A1-B2.
Thank you
With Reagrds
Kavya
----------
From: Vellieux Frederic
Hi,
If, in your case, no possible asymmetric unit can contain A1-B2, then you deposit A1-B1 (or I suppose A2-B2...) and indicate to the PDB (like placing cards in the header cards) the operator to be used (and the subunit it applies to) in order to generate the most likely biological dimer. Normally the PDB can take care of that.
Fred.
Kayashree M wrote:
----------
From: Kayashree M
Thank you Sir for the suggestions.
The protein looks like the letter "C", So in one of the trimer it is arranged
as "C" while in the other trimer (stacked) it is arranged like "inverted C", So
The dimer A1-B1 and A2-B2 are same while A1-B1 and A1-B2 are different.
Thanking you
With Regards
Kavya
as "C" while in the other trimer (stacked) it is arranged like "inverted C", So
The dimer A1-B1 and A2-B2 are same while A1-B1 and A1-B2 are different.
Thanking you
With Regards
Kavya
----------
From: Eleanor Dodson
I am sure someone has suggested this - you can submit your coordinates to the PISA server which will give you a detailed analysis of contacts, then you choose which grouping you think makes the most reasonable assemblyn and take the asymmetric unit from that .
I actually like to do that while still refining - it makes coot a lot easier to use if you are working with a compact molecule!
Eleanor
----------
From: Kayashree M
Respected Madam,
Actually I was not aware of the fact that we have to deposit the structure which is most probable
biological assembly (since in PDB there are seperate representations for the asymmetric unit and
biological unit).
Also Since I dint have any experimental evidences other than PISA predictions and comparison
with the structures from different organisms, I continues to refine the model PHASER predicted.
In addition the PHASER refined model has 3 Cadmium ions inbetween the monomers strongly
coordinated by both the monomers. Hence I did not have any doubts upon this assembly. Finally
I submitted this assembly itself. and by mistake I mentioned most probably biological assembly
section while submitting the PDB, Which gave rise to all confusions.
Thanking you
With Regards
M. Kavyashree
----------
From: <eugene.krissinel
Dear Bostjan,
Not at all. But if anticipated biological assembly has the same size as ASU,
then one should be able to choose ASU such that it is biological assembly.
Consider that, when there is more than 1 chain in ASU, its configuration
may be chosen in many different ways (but not completely arbitrarily,
of course). E.g., in case of heterodimeric ASU, _any_ hetero-pair of molecules
in crystal is a valid ASU (even if they do not make a contact :)). By this I mean
that, by applying all crystal symmetry operations and unit cell translations,
one can reconstruct the whole crystal from an arbitrary hetero-pair. In case
of homodimers it is slightly more complex, e.g. one has to exclude
monomers in parallel orientations.
I would think (am I wrong here?) that NCS-reduced PDB entries do not
represent crystal's ASU (simply because they are reduced by
_non-crystallographic_ symmetry operations). In order to get the ASU,
one needs to apply NCS to the content of the entry. E.g., PDB entry
1stm has 5 chains, but they do not make an ASU: applying all space
group symmetry operations would not reconstruct the crystal.
NCS-expansion, however, gives a 60-chain viral capsid, which is the
ASU. For fun, you may state that "your" ASU in this case is 2
complementing parts of neighbouring capsids, and this would be
"crystallographically correct". It is a matter of common sense (and
sense of beauty perhaps) that we choose to deposit a complete
viral capsid as an ASU here.
Eugene.
----------
From: James Stroud
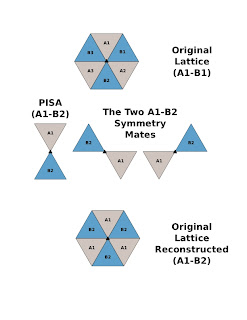
Since a crystallographic 3-fold generates the trimer (of dimers) then A1-B2 can be the ASU in this case.
Just generate the symmetry mates with the A1-B1 dimer and then make a new PDB of A1-B2 then generate the symmetry mates of A1-B2 to see that the lattice is complete.
I compulsively made and attached an illustration to show what I mean.
James

----------
From: Kayashree M
Respected Sir,
Thank you for the efforts and nice depiction of the problem.
The Dimeric assembly that is solved and the one predicted
by PISA is depicted in the attachment. This is the arrangement
I wanted to describe. The protein overall appears like a "C".

Thank you for the efforts and nice depiction of the problem.
The Dimeric assembly that is solved and the one predicted
by PISA is depicted in the attachment. This is the arrangement
I wanted to describe. The protein overall appears like a "C".
Since a crystallographic 3-fold generates the trimer (of dimers) then A1-B2 can be the ASU in this case.

No comments:
Post a Comment